






 Author Affiliations
Author Affiliations
Abstract
Primary neuroendocrine tumors of the ovary are a heterogeneous family of neoplasms that have a wide range of characteristics and prognosis. Relatively little research has been done on the topic of genetic commonalities in this class. The authors sampled four cases of neuroendocrine ovarian tumors, two well-differentiated neuroendocrine tumors and two non-small cell neuroendocrine tumors. Deoxyribonucleic acid (DNA) libraries were prepared, and 14 genes of interest were found in one or more of the cases. Six genes of interest were identified in all four cases: Cancer antigen 125 (CA125), Mucin 6 (Muc6), tumor protein 53 (TP53), ataxia telangiectasia mutated (ATM), lysine methyltransferase 2D (KMT2D), and neurotrophic tyrosine receptor kinase 1 (NTRK1).
Keywords
Whole exome sequencing, Neuroendocrine neoplasms, Deoxyribonucleic acid, Genes, Non-small cell neuroendocrine tumors.
Introduction
Neuroendocrine tumors of the ovary consist of a group of heterogeneous malignancies that vary in biological behavior and prognosis. In recent years, several new molecular markers and diagnostic tests for the early detection of these tumors have been investigated. However, these studies have been based on tumors originating in various anatomic sites.[1] Data on the biomarkers of neuroendocrine malignancies of ovarian origin are limited.[2] Unspecific immunohistochemical stains such as neural cell adhesion molecule 1 (CD56), synaptophysin, chromogranin A, and c-kit are standard methods of confirming diagnosis. Electron microscopy is another, though less established, diagnostic pattern.
Genetic characterization is helpful for diagnosis and is becoming increasingly essential in treatment. The heterogeneous nature of this group is a primary reason. For example, while neuroendocrine neoplasms associated with ovarian teratomas appear to have good prognostic value and are easily treated, others like ovarian non-small cell neuroendocrine tumors tend to be found in advanced stages and have a poor prognosis.[3,4] With overlapping morphologic features, light microscopic diagnosis can be challenging. This is to say nothing of treatment, which does not currently offer an agreed-upon guideline. Only superficial consensus has been reached, agreeing that low stage and low grade offer better prognosis, with hysterectomy +/- salpingo-oophpherectomy variants is the standard surgical approach.[5,6] This should further embolden the medical community to search for genetic answers to these neoplasms.
In this paper, we report on the genomic aberrations of well-differentiated and poorly differentiated primary ovarian neuroendocrine neoplasms. It is the authors’ hope that this will be a step toward initiating larger-scale research to identify common genes and pathways within this family of cancers.
Methodology
Sample tumor DNA was collected from formalin-fixed paraffin-embedded tissue of four cases after review by H&E. Two of these cases were well-differentiated neuroendocrine tumors, and two were non-small cell neuroendocrine carcinomas (Table 1). DNA libraries were prepared using Illumina’s Nextera Rapid Capture Expanded Exome Kit, and the pair-end sequencing was performed on Illumina’s Nextseq 500 system with Nextseq 500 high-output kit (150 cycles). Read alignment and variant cell analyses were then performed with Illumina’s Burrows-Wheeler aligner (BWA), Enrichment, VariantStudio, and NextGene software.
| Diagnosis |
| 1. Non-small cell, neuroendocrine undifferentiated carcinoma |
| 2. Mucinous carcinoma (monodermal teratoma) |
| 3. Non-small cell neuroendocrine carcinoma of the ovary |
| 4. Neuroendocrine teratoma of the ovary, benign cystic teratoma (dermoid cyst) |
Table 1: Summary of cases selected for publication
Results
By BWA Enrichment analysis, we identified 32,519 – 45,523 single-nucleotide variants (SNV), 698 – 1,358 insertion variants, and 830 – 1,650 deletion variants. By VariantStudio, these were filtered by passing criteria of read depth (30x), frequency (5%), and consequence. Missense variants were 2,847 to 5,961, frameshift truncations were 14 to 41, and variants to a stop again were 21 to 54. Comparative assessment by NextGene demonstrated that there were 202 missense or frameshift mutations in 141 genes shared among all four cases. Additional mutations in epidermal growth factor receptor (EGFR), TP53, NTRK1, KMT2D, tuberous sclerosis complex 1 (TSC1), and phosphatase and tensin homolog (PTEN) were unique to certain cases of well and poorly differentiated neoplasms.
Missense mutations of CA125, Muc6, TP53, ATM, KMT2D, and NTRK1 were present in all four cases. All of these were SNVs with 11 different amino acids involved in the variants. 5 different chromosomes were involved, though chromosome 11 appeared twice with the Muc6 and ATM genes.
When also considering genes that were present in 3 or less of the cases, the vast majority of cases are still SNVs as well as missense mutations. No clear pattern is present of chromosomes involved, though 3 genes are on 17. Genotypes are homogenous, heterogenous, or both, with 8 of the 14 genes being purely heterozygous (Table 2).
| Gene | No. of cases | Chromosome | Type | Consequence | Genotype | Variant |
| CA125 | 4 | 19 | SNV | Missense | Homozygous | Valine (Val) 14446 Leucine (Leu) |
| Muc6 | 4 | 11 | SNV | Missense | Heterozygous | Tyrosine (Tyr) 1826 Aspartic acid (Asp) |
| TP53 | 4 | 17 | SNV | Missense | Homo/Hetero | Proline (Pro) 72 Arginine (Arg) |
| MYH | 3 | 17 | SNV | Missense | Homo/Hetero | Tryptophan (Trp) 5074 Arginine (Arg) |
| EGFR | 2 | 7 | SNV | Missense | Heterozygous | Arginine (Arg) 521 Lysine (Lys) |
| MutL Homolog 1 (MLH1) | 2 | 3 | SNV | Missense | Homo/Hetero | Isoleucine (Ile) 219 Valine (Val) |
| ATM | 4 | 11 | SNV | Missense | Homozygous | Asparagine (Asn) 1983 Serine (Ser) |
| Neurofibromin 1 (NF1) | 1 | 17 | SNV | Missense | Heterozygous | Aspartic acid (Asp) 176 Glutamic acid (Glu) |
| TSC1 | 1 | 9 | SNV | Missense | Heterozygous | Methionine (Met) 322 Threonine (Thr) |
| KMT2D | 4 | 12 | SNV | Missense | Heterozygous | Alanine (Ala) 476 Threonine (Thr) |
| NTRK1 | 4 | 1 | Snv | Missense | Heterozygous | Threonine (Thr) 237 Methionine (Met) |
| Melanoma Antigen Family C1 (MAGEC1) | 1 | X | SNV | Missense | Heterozygous | Alanine (Ala) 229 Proline (Pro) |
| PTEN | 3 | 10 | Deletion | Truncation | Homozygous | ——- |
| Phosphatidylinositol-4,5-bisphosphate 3-Kinase Catalytic Subunit Alpha (PIK3CA) | 3 | 3 | SNV | Missense | Heterozygous | Histidine (His) 1047 |
Table 2: Genes present in cases selected
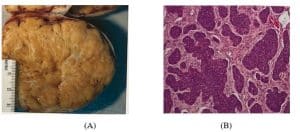
Figure 1: Case 4
- Gross section of tumor reveals a firm, yellow, solid tumor that involves the entire ovarian parenchyma. This is somewhat smaller and more solid than is usually expected for this type of tumor
- H&E microscopic section showing tumor growing in nests and trabeculae (x100)
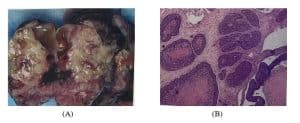
Figure 2: Case 3
- Gross section of tumor reveals a fleshy, tan tissue with focal hemorrhage, necrosis, and cystic change
- H&E microscopy reveals a tumor that grows as wide trabeculae or as solid sheets with areas of extensive necrosis (x100)
Discussion
Ovarian neuroendocrine tumors have historically been difficult to categorize. While their rarity is a contributing factor, there are other reasons as well. The true origin may not always be known, as neuroendocrine tumors of the midgut may metastasize to the ovaries in as many as 25% of female cases.[7] In teratomas, which are a heterogeneous group of neoplasms, the cells of origin may vary. Even in neuroendocrine tumors arising in the ovaries, teratomas may have any number of other cell lineages, including gastrointestinal, squamous, thyroid, and respiratory, making delineation of true origin ambiguous.[8] This study’s sample size was small, but one such tumor was just such an ovarian neuroendocrine teratoma, which in the context of scare research begs consideration.
While not present in this case series, another type of ovarian neuroendocrine tumor is the large cell neuroendocrine carcinoma. Their presentation is equally exotic, ranging from mucinous borderline to endometrioid adenocarcinoma to mucinous adenoma/cystadenoma and even unspecified types. Despite typically early staging, median survival is usually 8 months.[9]
Large proportions of neuroendocrine tumors can go misdiagnosed.[10] Immunohistochemistry has its limits. Though CD56, synaptophysin, chromogranin A, and c-kit are reliable and should be utilized if there is clinical suspicion, there are a variety of situations where they may fall short. These include occult origin, poor differentiation being common, and even frequent misdiagnosis of well-differentiated specimens.[11]
Despite all this, pathologists should not be discouraged, rather informed by infrequent, but invaluable research studies that are helping define this oncologic niche. The references in this paper are a suitable starting point for understanding the nuances of ovarian neuroendocrine neoplasms. Advances are being made, with new stains, such as insulinoma-associated protein 1 (INSM1), helpful in general diagnosis, and localized stains like lung neuroendocrine tumors (OTP) and midgut neuroendocrine tumors (CDX2), differentiating origin.[11] Furthermore, the WHO continues to develop new concepts to better differentiate and categorize neuroendocrine tumors in general, such as the mixed neuroendocrine, non-neuroendocrine, and amphicrine.[12]
Conclusion
Genomic abnormalities affecting multiple signaling pathways, including cell cycle progression, proliferation, and migration, as well as repair of DNA damage, are present in primary neuroendocrine neoplasms of the ovary. Most frequently recurrent abnormalities occur in the mucin gene family. Of note, a number of mutations encountered in this study have been reported to play a role in various human malignancies such as medulloblastoma and papillary thyroid carcinoma. The data reported herein may help contribute to the broader effort for a better understanding of the molecular aberrations that drive neuroendocrine carcinogenesis. Rather than being frustrated, we encourage pathologists to enthusiastically take on the challenge of piecing together the puzzle that neuroendocrine ovarian tumors present, as a means to sharpen diagnostic skills and enhance clinician experience.
References
- Szybowska M, Mete O, Weber E, Silver J, Kim RH. Neuroendocrine neoplasms associated with germline pathogenic variants in the homologous recombination pathway. Endocr Pathol. 2019;30(3):237-245. doi:10.1007/s12022-019-9569-4 PubMed | Crossref | Google Scholar
- Modlin IM, Bodei L, Kidd M. Neuroendocrine tumor biomarkers: From monoanalytes to transcripts and algorithms. Best Pract Res Clin Endocrinol Metab. 2016;30(1):59-77. doi:10.1016/j.beem.2016.01.002
PubMed | Crossref | Google Scholar - Opalińska M, Sowa-Staszczak A, Olearska H, et al. Clinical approach to neuroendocrine neoplasm associated with ovarian teratoma. Front Endocrinol (Lausanne). 2021;12:770266. doi:10.3389/fendo.2021.770266
PubMed | Crossref | Google Scholar - Veras E, Deavers MT, Silva EG, Malpica A. Ovarian nonsmall cell neuroendocrine carcinoma: a clinicopathologic and immunohistochemical study of 11 cases. Am J Surg Pathol. 2007;31(5):774-782. doi:10.1097/01.pas.0000213422.53750.d1 PubMed | Crossref | Google Scholar
- Zhu Y, Meng F, Fang H, et al. Clinicopathologic characteristics and survival outcomes in neuroendocrine carcinoma of the ovary. Int J Gynecol Cancer. 2020;30(2):207-212. doi:10.1136/ijgc-2019-000746
PubMed | Crossref | Google Scholar - Pang L, Guo Z. Primary neuroendocrine tumors of the ovary: Management and outcomes. Cancer Med. 2021;10(23):8558-8569. doi:10.1002/cam4.4368 PubMed | Crossref | Google Scholar
- Mulders MCF, de Lussanet de la Sablonière QG, van Velthuysen MLF, et al. Unfavorable biological behavior and treatment response of neuroendocrine ovarian metastases of midgut neuroendocrine tumors. Endocr Relat Cancer. 2023;30(8):e230035. doi:10.1530/ERC-23-0035 PubMed | Crossref | Google Scholar
- Leclerc J, Tihy M, Genestie C, et al. Updated morphological and immunohistochemical profile of neuroendocrine tumors developing in ovarian teratomas: A large series of a rare and heterogeneous disease. Int J Surg Pathol. 2024. doi:10.1177/10668969241271923 PubMed | Crossref | Google Scholar
- Burkeen G, Chauhan A, Agrawal R, et al. Gynecologic large cell neuroendocrine carcinoma: A review. Rare Tumors. 2020;12:2036361320968401. doi:10.1177/2036361320968401 PubMed | Crossref | Google Scholar
- Flores Legarreta A, Saab R, Gonzales NR, et al. Neuroendocrine neoplasms of the ovary: A review of 63 cases. Int J Gynecol Cancer. 2024;34(4):566-573. doi:10.1136/ijgc-2023-005063 PubMed | Crossref | Google Scholar
- Bellizzi AM. Immunohistochemistry in the diagnosis and classification of neuroendocrine neoplasms: What can brown do for you? Hum Pathol. 2020;96:8-33. doi:10.1016/j.humpath.2019.12.002 PubMed | Crossref | Google Scholar
- Rindi G, Mete O, Uccella S, et al. Overview of the 2022 WHO classification of neuroendocrine neoplasms. Endocr Pathol. 2022;33(1):115-154. doi:10.1007/s12022-022-09708-2 PubMed | Crossref | Google Scholar
Acknowledgments
Not reported
Funding
Not reported
Author Information
Corresponding Author:
Dominik Dabrowski
Department of Psychiatry
Rutgers University, New Jersey, USA
Email: dabrowskidom@gmail.com
Co-Authors:
Min Dai, Xinggui Shen, Hong Yin, Nestor Dela Cruz
Department of Pathology and Translational Pathobiology
Louisiana State University Health Science Center, Shreveport, USA
Eric Wei
Department of Pathology
University of South Alabama, USA
Authors Contributions
All authors contributed to the conceptualization, investigation, and data curation by acquiring and critically reviewing the selected articles. They were collectively involved in the writing – original draft preparation, and writing – review & editing to refine the manuscript. Additionally, all authors participated in the supervision of the work, ensuring accuracy and completeness. The final manuscript was approved by all named authors for submission to the journal.
Ethical Approval
All specimens were de-identified, and no patient data was shared beyond clinical presentation.
Conflict of Interest Statement
The author declares no conflict of interest.
Guarantor
None
DOI
Cite this Article
Dominik D, Min D, Xinggui S, Hong Y, Eric W, Nestor DC. Whole Exome Sequencing of Neuroendocrine Neoplasms of Ovarian Origin. medtigo J Med. 2025;3(1):e30623122. doi:10.63096/medtigo30623122 Crossref













