







 Author Affiliations
Author Affiliations
Abstract
Gaucher Disease (GD) is a rare, inherited lysosomal storage disorder resulting from a deficiency of the enzyme glucocerebrosidase. This deficiency leads to the accumulation of glucocerebroside in macrophages, causing a variety of systemic manifestations. This case report details the presentation, diagnosis, and management of a pediatric patient with Type 1 Gaucher Disease. The patient, a five-year-old male, exhibited symptoms including hepatosplenomegaly, anemia, and thrombocytopenia. Diagnostic confirmation was achieved through bone marrow biopsy and enzyme assay, revealing significantly reduced glucocerebrosidase activity. Management included enzyme replacement therapy (ERT), which resulted in significant clinical improvement. This report underscores the importance of early recognition and intervention in Gaucher Disease to prevent irreversible complications and improve patient outcomes. Additionally, it highlights the need for ongoing research and development of therapeutic options to enhance the quality of life for affected individuals.
Keywords
Lysosomal Storage Disorder, Gaucher Disease (GD), GBA1 gene, Glucocerebrosidase, Gaucher cells, Anemia, Thrombocytopenia, Hepatosplenomegaly
Introduction
Gaucher disease (GD) is a rare, genetic, autosomal recessive lysosomal storage disorder. The disease is named after the French physician Philippe Gaucher, who originally described it in 1882 [1]. The disease is characterized by the deficiency of the lysosomal enzyme glucocerebrosidase, encoded by the GBA1 gene, located on chromosome 1 (1q21) [2]. Glucocerebrosidase is responsible for hydrolyzing glucosylceramide into ceramide and glucose [3]. The deficiency of glucocerebrosidase leads to the accumulation of glucosylceramide in macrophages, transforming them into Gaucher cells [3]. Gaucher cells infiltrate various organs, primarily the bone marrow, spleen, and liver [3]. These cells have a distinctive appearance, described as “crumpled tissue paper”, with an eccentric nucleus, condensed chromatin, and enlarged cytoplasm [3]. The diagnosis of GD is based on clinical suspicion and confirmed by enzyme activity assays, which measure the activity of glucocerebrosidase in blood samples [3]. The buildup of glucocerebroside initiates a series of eventual consequences, such as inflammation, oxidative stress, and disruption of cellular functions. This contributes to the extensive involvement of multiple systems that are observed in Gaucher’s disease. For example, the invasion of Gaucher cells into the bone marrow hinders the normal production of blood cells, resulting in anemia and thrombocytopenia. The buildup in the bones impairs bone remodeling, which results in pathological fractures, avascular necrosis, and osteopenia [3]. Across all types of Gaucher disease, incidence estimates range from 0.45 to 25.0 per 100,000 live births. The lowest incidence is observed in the Asia-Pacific region. Type-specific prevalence estimates per 100,000 population are as follows: GD1: 0.26–0.63, GD2 and GD3: 0.02–0.08 (limited data from Europe) and Unspecified or overall GD: 0.11–139.0 (highest in North America) [4].
Case Presentation
A 5-year-old male child was brought by his parents with complaints of pain in the abdomen associated with progressive abdominal distension for the last six months. The patient also complained of fatigue and easy bruising. He had a significant family history wherein another sibling was treated for similar symptoms and deceased at the age of three due to worsening of the condition despite several blood transfusions. Both parents denied having any major significant medical history of their own. On examination, the child showed severe pallor and pedal edema. On abdominal palpation, he had splenomegaly as well as hepatomegaly. Neurological examination was within normal limits.
Case Management
Laboratory investigation revealed a hemoglobin level of 5.5 g/dL, a white blood cell counts of 15900/mL, a platelet counts of 80000/mcL, and a reticulocyte count of 2.3%. Other biochemical parameters, including kidney function, liver function, and serum electrolytes, were within normal range. Abdominal ultrasound showed hepatosplenomegaly. A bone marrow aspiration was performed, which revealed the presence of crinkled paper macrophages in the marrow space as shown in figure 1. A beta-glucosidase test was also recommended, and the levels were found to be 6.2 nmol/h/mg.
The patient was advised to undergo enzyme replacement therapy.
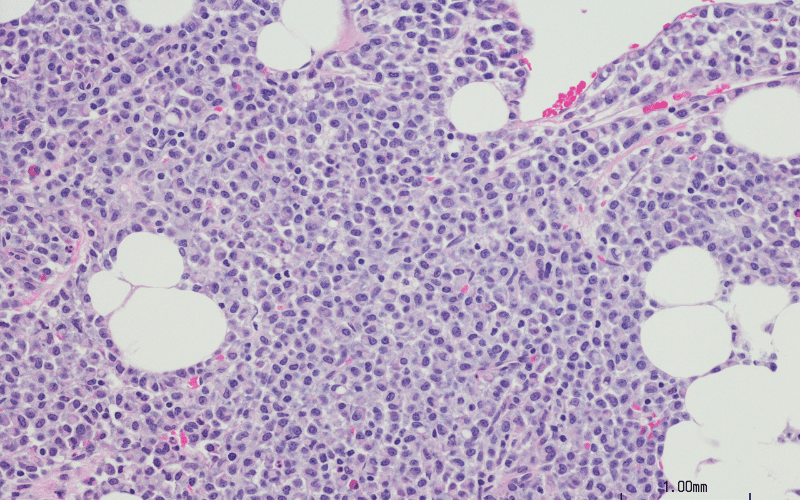
Figure 1: Bone marrow biopsy
Discussion
Gaucher’s disease is a condition characterized by a lack of the enzyme glucocerebrosidase, resulting into a dysfunction in intracellular storage chambers called lysosomes. This enzyme deficit leads to the buildup of glucocerebroside in several tissues of the body, with a specific impact on the spleen, liver, and bone marrow. The condition presents itself in three main forms: Type 1 (non-neuropathic), Type 2 (acute neuropathic), and Type 3 (chronic neuropathic) [5]. It is highly prevalent among the Ashkenazi Jewish people, particularly for Type 1. Type 1 is the most common form of Gaucher’s disease in children. Pediatric instances, particularly Types 2 and 3, are less common but have a higher severity and are associated with considerable morbidity and death. The condition is inherited in an autosomal recessive way, requiring the presence of a defective gene in both parents [5]. Gaucher’s disease is caused by mutations in the GBA gene, which is responsible for producing the enzyme glucocerebrosidase. More than 300 mutations have been discovered, spanning from single point mutations to intricate rearrangements. The predominant variants seen in the Ashkenazi Jewish population are N370S and L444P, with N370S being specifically associated with Type 1 and L444P being more frequently connected to Types 2 and 3. These genetic changes lead to different levels of enzyme insufficiency, which affects the intensity and characteristics of the disease [5].
Children diagnosed with Gaucher’s disease frequently exhibit hematological abnormalities, including anemia and thrombocytopenia. These abnormalities are caused by the invasion of Gaucher cells in the bone marrow. These symptoms may result in fatigue, increased susceptibility to infections, and a tendency for bleeding. Anemia is typically normocytic and normochromic, whereas thrombocytopenia can be severe, resulting in easy bruising and epistaxis [6,7,8]. Hepatosplenomegaly is a hallmark of Gaucher’s disease in pediatric patients. This can cause abdominal discomfort, early satiety, and, in severe cases, impair respiratory function due to the enlarged organs. The spleen can become massively enlarged, sometimes weighing several kilograms, and liver enlargement can contribute to a distended abdomen and altered metabolism [9]. Bone involvement in Gaucher’s disease leads to a range of complications, including bone pain, osteopenia, pathological fractures, and the characteristic Erlenmeyer flask deformity of the distal femur. These skeletal issues significantly impact the quality of life and physical development in pediatric patients. Bone crises, episodes of severe bone pain often accompanied by fever, are particularly debilitating and require prompt medical attention [10].
The diagnosis of Gaucher’s disease in pediatric patients involves a combination of clinical evaluation, biochemical tests, and genetic analysis. Enzyme assays measuring glucocerebrosidase activity in leukocytes or fibroblasts confirm the diagnosis. Genetic testing identifies mutations in the GBA gene, providing a definitive diagnosis and allowing for carrier screening within families. Early diagnosis is crucial for initiating appropriate treatment and improving outcomes. Prenatal diagnosis is possible through chorionic villus sampling or amniocentesis, allowing for early intervention and family planning. Newborn screening programs for lysosomal storage disorders are also being explored to facilitate early detection and treatment [11]. Niemann-Pick Disease, Sickle Cell Disease, Thalassemia, Chronic Myeloid Leukemia (CML), Juvenile Idiopathic Arthritis (JIA), Other Lysosomal Storage Disorders, Leukemia and Lymphoma, Osteomyelitis, Infectious Diseases, and Autoimmune Disorders are some conditions that may have symptoms similar to Gaucher’s disease in children [11].
Enzyme Replacement Therapy i.e. ERT is the mainstay of treatment for Gaucher’s disease. It involves the intravenous administration of recombinant glucocerebrosidase, which helps reduce the burden of accumulated glucocerebroside. ERT is particularly effective in managing hematological and visceral symptoms and improving quality of life. However, it has limited efficacy in treating the neurological manifestations of Types 2 and 3. ERT has transformed the management of Gaucher’s disease, leading to significant improvements in anemia, thrombocytopenia, hepatosplenomegaly, and bone pain. Regular infusions, typically every two weeks, are required to maintain therapeutic benefits [12].
Substrate Reduction Therapy i.e. SRT aims to decrease the production of glucocerebroside, thereby reducing its accumulation. It serves as an adjunct or alternative to ERT, particularly for patients who cannot tolerate ERT. SRT has shown efficacy in managing systemic symptoms but also has limited effects on neurological symptoms [13]. Two SRT agents, miglustat and eliglustat, are currently approved for Gaucher’s disease. Miglustat, an oral medication, inhibits the enzyme glucosylceramide synthase, reducing the synthesis of glucocerebroside. Eliglustat, a more selective inhibitor, has shown better tolerability and efficacy in clinical trials [13].
Gene Therapy or HSCT involves the transplantation of hematopoietic stem cells from a healthy donor, which can produce normal glucocerebrosidase and reverse the disease process. Advances in conditioning regimens and supportive care have improved outcomes, but the procedure remains limited by donor availability and risks [13].
Nutritional support and physical therapy are important components of comprehensive treatment. Adequate nutrition is necessary to support growth and development, while physical therapy helps maintain mobility and reduce pain. Psychological support is also essential, as chronic illness can impact mental health and quality of life [12,13]. Along with all the above-mentioned components of management, supportive care is essential in managing Gaucher’s disease in pediatric patients. This includes regular monitoring of growth and development, pain management, and treatment of complications such as anemia and fractures. Multidisciplinary care involving pediatricians, hematologists, neurologists, and genetic counselors is crucial for comprehensive management [14].
The prognosis of Gaucher’s disease varies depending on the type and severity of the disease. Type 1 patients typically have a normal life expectancy with appropriate treatment. In contrast, types 2 and 3 are associated with significant morbidity and reduced life expectancy. Early diagnosis and intervention are key to improving outcomes, particularly in pediatric patients [15].
Ongoing research and advances in treatment have improved the outlook for Gaucher’s disease. Long-term follow-up studies have demonstrated the benefits of ERT and SRT in reducing disease burden and improving survival. However, challenges remain in managing neurological symptoms and optimizing treatment regimens [15].
Recent research has focused on developing novel therapies targeting the neurological manifestations of Gaucher’s disease. Gene therapy, which aims to correct the underlying genetic defect, and pharmacological chaperones, which enhance the residual activity of mutant glucocerebrosidase, are promising areas of investigation. Additionally, research into the pathophysiological mechanisms linking Gaucher’s disease to Parkinson’s disease has provided insights into potential therapeutic targets [15]. Gene therapy approaches, such as the use of adeno-associated virus vectors to deliver functional copies of the GBA gene, have shown promise in preclinical studies. Clinical trials are underway to evaluate the safety and efficacy of these approaches. Pharmacological chaperones, small molecules that stabilize misfolded glucocerebrosidase, have also shown potential in enhancing enzyme activity and reducing substrate accumulation [15].
Due to the presence of various clinical differences, patients frequently receive incorrect diagnoses and encounter significant challenges in obtaining a precise diagnosis. Survey data reveals that approximately 16% of patients have a delay of at least 7 years before receiving a diagnosis of GD following their initial consultation with a doctor. Delays in diagnosis may occur due to a lack of knowledge of GD or incorrect identification, the presence of varied physical characteristics or non-specific symptoms, the presence of mild symptoms, and the use of external testing services. Incorrect diagnosis can result in worsening of the disease, continued presence of untreated symptoms, emotional anguish, and feelings of frustration and melancholy due to the absence of an explanation for the symptoms [15].
Conclusion
In conclusion, this case report emphasizes the critical need for early diagnosis and intervention in Gaucher Disease, particularly in pediatric patients. The presented case of a five-year-old male patient with Type 1 Gaucher Disease demonstrates the effectiveness of enzyme replacement therapy (ERT) in alleviating symptoms and improving clinical outcomes. Early identification and treatment can significantly reduce the risk of severe complications, enhance quality of life, and promote a better long-term prognosis. This report also highlights the importance of genetic testing and enzyme assays in confirming the diagnosis of Gaucher Disease. Ongoing research and advancements in therapeutic options are essential to further improve the management of this condition and to offer better prospects for affected individuals.
References
- Gaucher E. De l’epithelioma primitif de la rate: hypertrophie idiopathique de la rate sans leucémie 2024. De l’epithélioma primitif de la rate
- Garça M, Correia S, Goulart A, Ávila P. Gaucher disease: One of the few causes of massive splenomegaly. Eur J Case Rep Intern Med. 2022;9(12):003705. doi.10.12890/2022_003705. PubMed | Crossref | Google Scholar
- Stirnemann J, Belmatoug N, Camou F, et al. A review of Gaucher disease pathophysiology, clinical presentation, and treatments. Int J Mol Sci. 2017;18(2):441 doi.10.3390/ijms18020441 PubMed | Crossref | Google Scholar
- Castillon G, Chang SC, Moride Y. Global incidence and prevalence of Gaucher disease: A targeted literature review. J Clin Med. 2022;12(1):85. doi:10.3390/jcm12010085. PubMed
- Stirnemann J. Gaucher disease: Review, epidemiology and modelling of the biological and clinical markers evolution (Privat-docent thesis). University of Geneva; 2016. Gaucher disease: Review, epidemiology and modelling of the biological and clinical markers evolution
- Özdemir GN, Gündüz E. Gaucher disease for hematologists. Turk J Hematol. 2022;39(2):136-139. doi.10.4274/tjh.galenos.2021.2021.0683 PubMed | Crossref | Google Scholar
- Huang WJ, Zhang X, Chen WW. Gaucher disease: A lysosomal neurodegenerative disorder. Eur Rev Med Pharmacol Sci. 2015;19(7):1219-1226. Gaucher disease: A lysosomal neurodegenerative disorder
- Shoham OZ. Gaucher disease in children (Master’s thesis). University of Zagreb, School of Medicine; 2018. Gaucher disease in children
- 9.Basilicata M, Marrone G, Di Lauro M, et al. Gaucher disease in internal medicine and dentistry. Appl Sci. 2023;13(4062). Gaucher disease in internal medicine and dentistry
- Katakam K. Overview of Gaucher disease and its management. World J Pharm Res. 2017;563-584. Overview of Gaucher disease and its management
- Gupta P, Pastores G. Pharmacological treatment of pediatric Gaucher disease. Expert Rev Clin Pharmacol. 2018;11(12):1183-1194. doi.10.1080/17512433.2018.1549486 PubMed
- Hughes DA, Pastores GM. Gaucher disease. In: Adam MP, Feldman J, Mirzaa GM, et al., eds. GeneReviews® (Internet). University of Washington, Seattle; 2000. Gaucher disease
- Kaplan P, Baris H, De Meirleir L, et al. Revised recommendations for the management of Gaucher disease in children. Eur J Pediatr. 2013;172(4):447-458. doi.10.1007/s00431-012-1771-z. PubMed | Crossref | Google Scholar
- Weinreb NJ, Goker-Alpan O, Kishnani PS, et al. The diagnosis and management of Gaucher disease in pediatric patients: Where do we go from here? Mol Genet Metab. 2022;136(1):4-21. doi.10.1016/j.ymgme.2022.03.001 PubMed | Crossref | Google Scholar
- Mehta A, Belmatoug N, Bembi B, et al. Exploring the patient journey to diagnosis of Gaucher disease from the perspective of 212 patients with Gaucher disease and 16 Gaucher expert physicians. Mol Genet Metab. 2017;122(3):122-129. doi.10.1016/j.ymgme.2017.08.002 PubMed | Crossref | Google Scholar
Acknowledgments
Not applicable
Funding
Not applicable
Author Information
Corresponding Author:
Sheetal Hiremath
Independent Researcher, Department of Content
medtigo India Pvt Ltd, Pune, India
Email: [email protected]
Co-Author:
T Sindhoori
Independent Researcher, Department of Content
medtigo India Pvt Ltd, Pune, India
Authors Contributions
All patient-related data were collected by Dr. Sindhoori. Dr. Sheetal Hiremath contributed to the writing of the manuscript, including the original draft preparation and subsequent review and editing to refine the final version.
Informed Consent
Not applicable
Conflict of Interest Statement
This case report is based on a clinical case published in the “Cases” section of medtigo.com. The authors declare no conflicts of interest related to this publication.
Guarantor
Not applicable
DOI
Cite this Article
T, S., & Hiremath, S. How Challenging to Diagnose and Treat Gaucher’s Disease in Children. medtigo J Med. 2024;2(2):e3062223. doi:10.63096/medtigo3062223 Crossref













