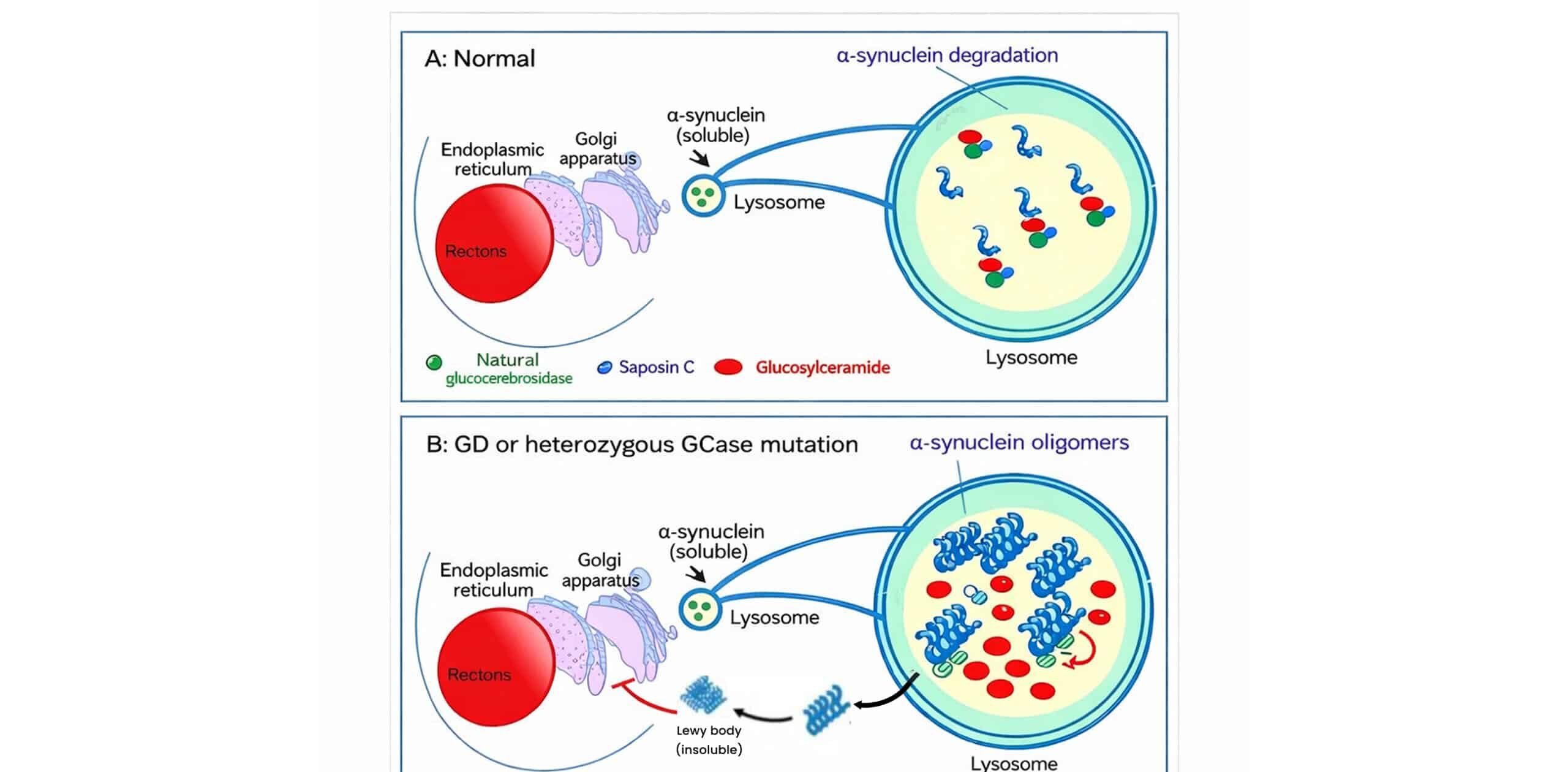
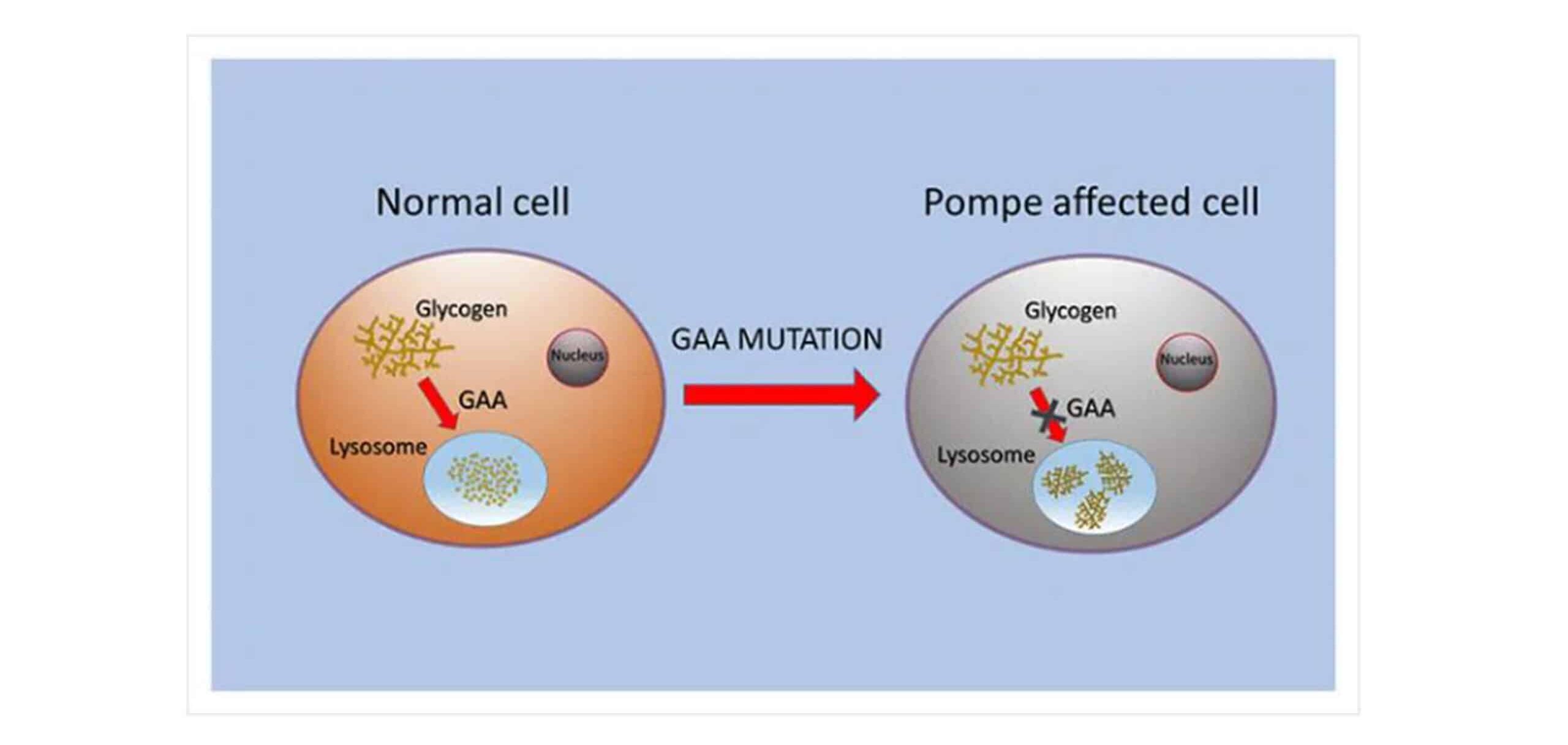
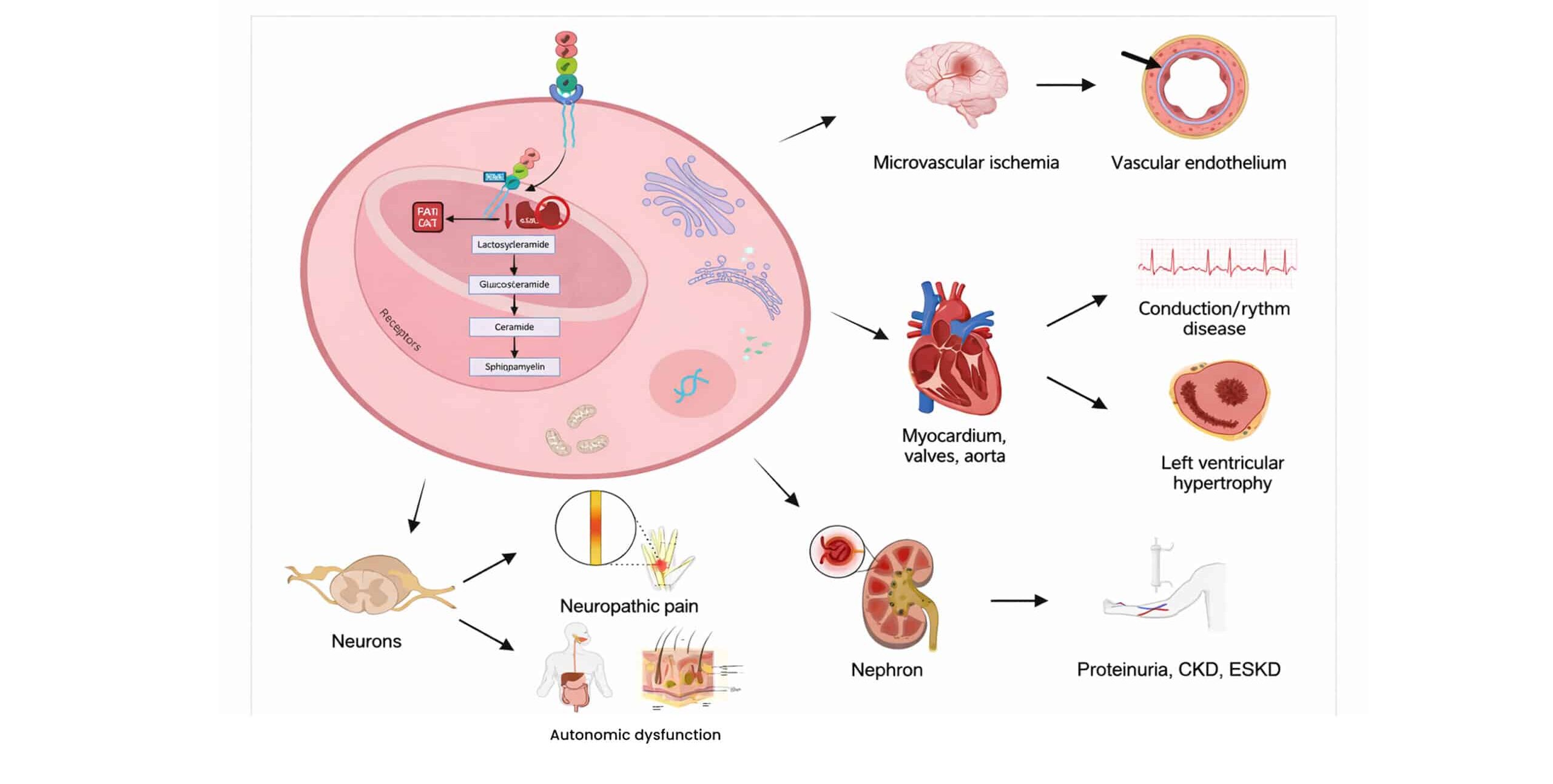
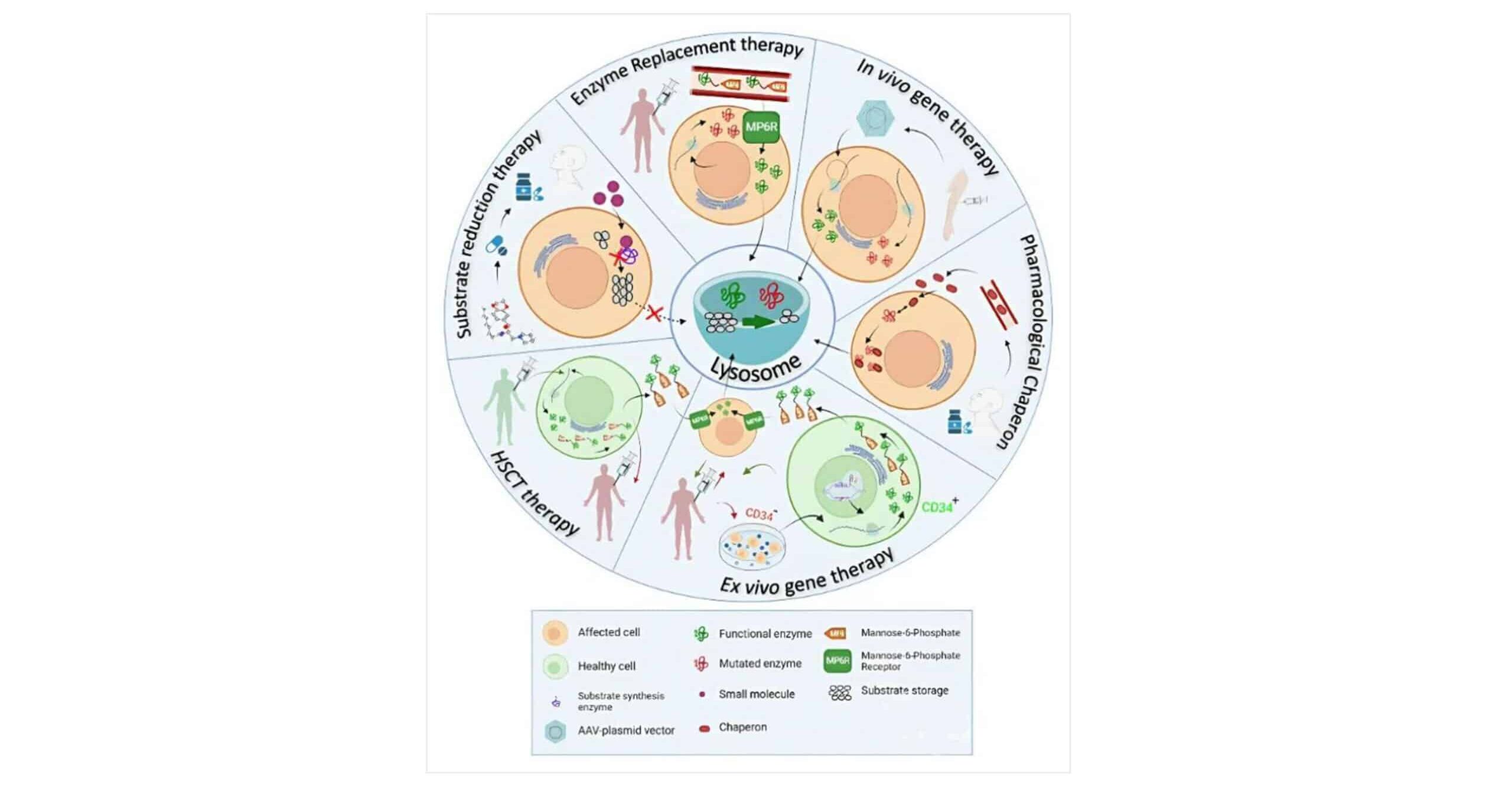
Author Affiliations
Author Affiliations
This article has been retracted in accordance with medtigo Journal of Pharmacology policy (https://journal.medtigo.com/journal-insights/#pub_inte_and_retra). In a post-publication review, concerns were identified regarding permission for use and the copyright status and of certain images used in the article. The author failed to respond to requests for confirmation of permission to use the images in their submission and in our publication. OR The author was unable to provide confirmation of permission to use the images in their submission and in our publication. As a result, the Administrative Editor of the medtigo Journal of Pharmacology has determined that retraction of this article is necessary to comply with ethical standards and to maintain the integrity of the scientific record.