







 Author Affiliations
Author Affiliations
Abstract
Creutzfeldt-Jakob disease (CJD) is an uncommon and fatal neurodegenerative disorder, typically affecting the elderly population. Its rarity and varied clinical presentation pose unique challenges in diagnosis. The case report under consideration involves an adult male, highlighting the importance of early diagnosis. Given its unusual nature and rapid onset, the application of exclusion criteria through a thorough differential diagnosis proves invaluable in confirming CJD. There is no specific treatment available to cure or halt the progression of this condition. The primary approach involves supportive treatment and counseling, aiming to enhance the quality of life for individuals affected by this challenging disorder. In this case report, we have discussed the case of a 61-year-old man with complaints of rapid cognitive decline leading to difficulty in activities of daily living. The MRI brain impression showed increased signal mostly in the caudate nuclei and, to a lesser extent, the putamen and thalami.
Keywords
Creutzfeldt-Jacob disease, Neurodegenerative, Cognitive, Prion disease, Transmissible spongiform encephalopathy (TSE), Prion proteins
Introduction
Speilmeyer first coined the term Creutzfeldt-Jacob disease in 1922 [1]. It was based on the names of Creutzfeldt and Jacob, two doctors who documented patients’ cases with unusual neurological symptoms associated with other conditions [1]. The diagnosis of Creutzfeldt-Jacob disease was confirmed only a few years later with the help of modern diagnostic techniques [2]. Creutzfeldt-Jakob Disease is a neurodegenerative disorder that is considered rare, progresses rapidly, and has a 100% fatality rate. Among the different forms of this disease, the most common type is the sporadic form. The primary challenge associated with CJD (Creutzfeldt-Jakob Disease) lies in its diagnosis. Although confirmation through histopathology is considered the yardstick for a definitive diagnosis, advancements in testing have led to the availability of less invasive pre-mortem diagnostic options, thereby reducing the need for brain biopsies. By incorporating imaging studies, electroencephalography, and biomarkers together with clinical observations, the diagnosis of CJD can be facilitated without the need to obtain brain tissue samples [3]. In the United States, approximately 350 cases of CJD are reported each year [4]. This case report aims to provide a clearer understanding of the presentation and management of patients with CJD.
Case Presentation
A 61-year-old male presented to the physician’s office, brought by his daughter, reporting symptoms indicative of cognitive decline and alterations in behavior. For the last six weeks, the patient has manifested pronounced somnolence, difficulties in name recall, and episodes of disorientation in his surroundings. His ability to perform daily activities, such as meal preparation and self-care, has been notably compromised. Additionally, he exhibits upper arm spasms when subjected to sudden stimuli. Pertinent medical history includes a diagnosis of diabetes mellitus managed with metformin and a substantial smoking history of 30 pack years. While there is no documented history of psychiatric conditions, the patient’s father was diagnosed with Alzheimer’s disease around the age of 60. Clinical examination revealed a body temperature of 98.9°F, blood pressure of 120/80 mm Hg, a pulse rate of 75/min, and a respiratory rate of 16/min. The patient remains alert and oriented, albeit with a Montreal Cognitive Assessment score of 15/30 (normal: ≥26). MRI was indicated and was taken for further evaluation. The brain MRI image of this patient is a DWI or diffusion-weighted image showing abnormal increased signal mostly in the caudate nuclei and, to a lesser extent, the putamen and thalami. The findings are symmetric. There is the deep grey matter, which can be seen in CJD as shown in Figure 1.
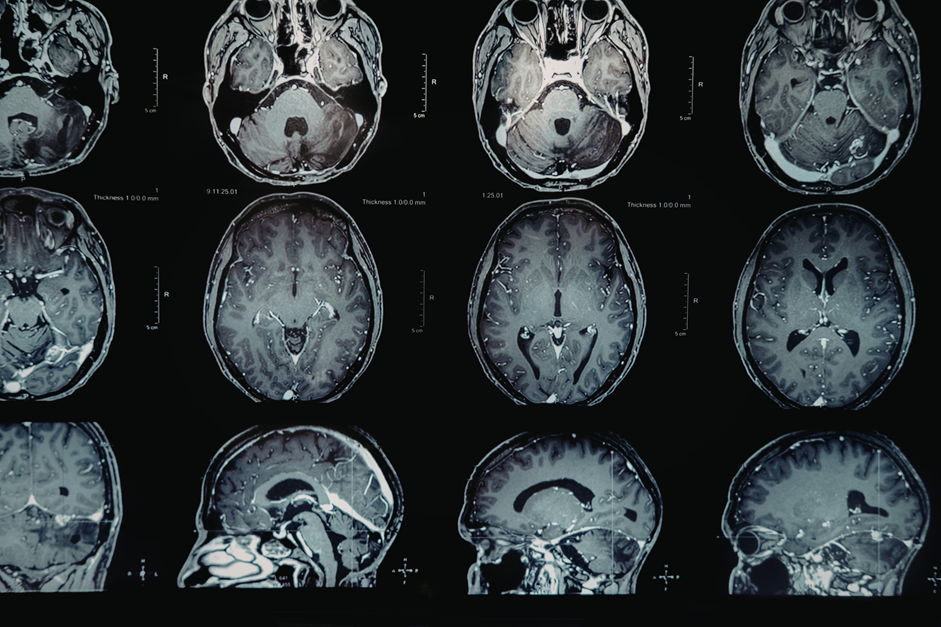
Figure 1: MRI Brain
Case Management
Even with the progress in comprehending this ailment, the outlook remains bleak, as CJD is ultimately fatal. The primary approach to treatment is symptomatic and supportive, such as employing clonazepam to address myoclonus [5].
The primary focus in managing CJD is to alleviate symptoms and enhance the individual’s comfort. Various measures can be implemented, including the use of medications such as antidepressants, pain relievers, and sedatives to address anxiety, depression, pain, and insomnia. Nursing care is provided, encompassing assistance with feeding, hygiene, and mobility. Creating a safe and serene environment is essential, characterized by sufficient lighting, familiar objects, and minimal noise. Emotional and practical support is extended to the individual, their family, and caregivers. Additionally, involvement in research studies or clinical trials that may offer new insights or treatments for CJD is encouraged.
In the specific case mentioned, the neurologist counseled and educated the patient and his family in the above-mentioned manner, providing supportive treatment.
Discussion
Pathophysiology
Prion diseases, also known as transmissible spongiform encephalopathies (TSEs), encompass a group of rare and destructive neurological disorders. Unlike other contagious agents, these disorders arise due to the misfolding of normal cellular proteins called prion proteins. Termed PrPSc, these abnormal prion proteins accumulate within the brain, consequently causing gradual harm and sponge-like alterations in neural tissue. Initially harmless, prion proteins naturally exist in the body and manifest as PrPC (C for cellular) in their normal cellular form. The misfolded transformation from PrPC to PrPSc triggers the pathological series of events in prion diseases. The buildup of misrepresented prion proteins eventually triggers the creation of amyloid plaques and neurofibrillary tangles in the brain, leading to a progressive breakdown of neurons, sponge-like modifications, and, ultimately, severe neurological dysfunction [3].
Types of CJD
- Sporadic CJD: The most prevalent form of the disease is the sporadic type known as sCJD. Typically, onset occurs during the seventh decade of life, and individuals with sCJD have a median survival time of five months, with 90% succumbing to the disease within one year. It shows an annual incidence of one case per one million individuals, constituting approximately 85% of all CJD cases. The agent responsible for causing the infection is the abnormal form called “scrapie” (PrPSc) of the prion protein found in the host’s cells (PrPC). This form induces a modification of PrPC into the disease form through posttranslational processes. This accumulation in the brain leads to neurodegeneration [3].
- Familial CJD: Mutations in prion proteins are responsible for familial CJD, constituting around 10% of all prion disease cases [6].
- Variant CJD: The term “Variant CJD” came into the picture in 1996 when it was understood that it is a result of eating food contaminated with bovine spongiform encephalopathy. It was a significant problem in the UK back then [7].
Signs and Symptoms
The physical or neurological examination findings differ slightly with the phenotypic strain. However, the patient shows common symptoms such as rapid cognitive decline, dementia of unknown origin, ataxia, and visual changes that manifest as cortical blindness, myoclonus, and akinetic mutism in the last stage of this disease. The most atypical symptoms are insomnia, psychiatric symptoms, chorea, and peripheral neuropathy [8].
Diagnosis
In 1998, the World Health Organization (WHO) introduced diagnostic criteria for CJD, where diagnosis was primarily based on clinical examination, EEG, and cerebrospinal fluid (CSF) findings. However, this approach might not work in modern medicine, as it does not consider MRI findings, genetic testing, or contemporary laboratory tests to confirm the diagnosis. The criteria include EEG and the assessment of the 14–3-3 protein in the CSF, both of which are usually conducted during the initial stages of the diagnostic evaluation [9]. Though in most cases, CSF analysis reveals elevated levels of 14-3-3 proteins, it is not considered very specific [9]. Most of the patients show periodic sharp wave complexes in EEG [9]. Typically, an MRI scan of a patient with CJD reveals increased signal intensity in the striatum or thalamus on
T2-weighted images. Additionally, lesions may be observed in the periventricular white matter and generalized cortical atrophy [9,10].
Differential Diagnosis
The neurological symptoms of the patient may mimic several types of dementia, including Alzheimer’s disease, frontotemporal dementia, vascular dementia, and Lewy body dementia. It is important to note that while all other conditions that cause dementia have a gradual onset, dementia associated with Creutzfeldt-Jakob disease has a rapid onset [10].
The other conditions that can be misdiagnosed are normal pressure hydrocephalus, Parkinsonian disorder, viral encephalitis, paraneoplastic disorder, depression, peripheral vertigo, stroke, central nervous system vasculitis, peripheral neuropathy, Hashimoto’s encephalopathy, infectious diseases, metabolic disorders, and any other autoimmune process [10].
Huntington’s disease is another neurodegenerative disorder that can be mistaken for CJD. However, Huntington’s disease has a strong family history. Generalized biphasic or triphasic sharp wave complexes in EEG cannot be seen in patients with Huntington’s disease. Also, CSF examination does not reveal the presence of 14-3-3 proteins [11]
One of the case reports describes a 70-year-old man who presented with anxiety and dementia for eight weeks and was diagnosed with sporadic CJD based on the 2017 Euro-CJD criteria. The key findings were cortical hyperintensity on diffusion-weighted MRI, periodic triphasic waves on EEG, and positive 14-3-3 protein in CSF [12].
A similar case report illustrates a 70-year-old patient who developed akinetic mutism after three months of behavioral changes and involuntary movements. He was diagnosed with probable sporadic CJD based on the WHO criteria. The key features were cortical ribboning on diffusion-weighted MRI, periodic triphasic waves on EEG, and positive 14-3-3 protein in CSF. This patient died 4 months after he started his complaints [13].
A case of sporadic CJD was examined in a 48-year-old woman who showed signs of depression, insomnia, anorexia, and silence that began two months prior to the first consultation. Her symptoms worsened over time. The findings that confirmed the diagnosis of the disease were vague neuropsychological symptoms, increased signal intensity in the caudate and putamen regions on brain MRI, widespread high-frequency spike waves on EEG, and detection of 14-3-3 protein in the spinal fluid. She received symptomatic treatment [14].
Conclusion
Creutzfeldt-Jakob disease is an exceedingly rare and peculiar condition that can present a challenge for healthcare providers. However, if a healthcare provider is well equipped with their knowledge and knows the diagnostic tests, then it could impact overall disease management. There might be a slight variation when a healthcare provider orders any test based on the type of this disease. In some cases, the knowledge of different strain phenotypes is necessary, while in others, brain biopsy can be performed.
RT-QuIC is one of the significant diagnostic tools. In the case mentioned above, the MRI brain proved to be the primary diagnostic tool that helped diagnose this patient.
References
- Henry R, Murphy FA. Etymologia: Creutzfeldt-Jakob Disease. Emerg Infect Dis. 2017;23(6):956. Crossref | Google Scholar
- Johnson RT, Gibbs CJ Jr. Creutzfeldt–Jakob disease and related transmissible spongiform encephalopathies. N Engl J Med. 1998;339(27):1994-2004. doi:10.1056/NEJM199812313392707 PubMed | Crossref | Google Scholar
- Manix M, Kalakoti P, Henry M, et al. Creutzfeldt-Jakob disease: updated diagnostic criteria, treatment algorithm, and the utility of brain biopsy. Neurosurg Focus. 2015;39(5):E2. doi:10.3171/2015.8.FOCUS15328 PubMed | Crossref | Google Scholar
- Creutzfeldt-Jakob disease. National Institute of Neurological Disorders and Stroke. Creutzfeldt-Jakob disease
- Sequeira D, Nihat A, Mok T, et al. Prevalence and treatments of movement disorders in prion diseases: a longitudinal cohort study. Mov Disord. 2022;37(9):1893-1903. doi:10.1002/mds.29152 PubMed | Crossref | Google Scholar
- Mead S. Prion disease genetics. Eur J Hum Genet. 2006;14(3):273-281. doi:10.1038/sj.ejhg.5201544 PubMed | Crossref | Google Scholar
- Will RG, Ironside JW, Zeidler M, et al. A new variant of Creutzfeldt-Jakob disease in the UK. Lancet. 1996;347(9006):921-925. doi:10.1016/s0140-6736(96)91412-9 PubMed | Crossref | Google Scholar
- Head MW. Human prion diseases: molecular, cellular and population biology. Neuropathology. 2013;33(3):221-236. doi:10.1111/neup.12016 PubMed | Crossref | Google Scholar
- Chohan G, Pennington C, Mackenzie JM, Andrews MM, Everington D, et al. The role of cerebrospinal fluid 14-3-3 and other proteins in the diagnosis of sporadic Creutzfeldt-Jakob disease in the United Kingdom: A 10-year review. J Neurol Neurosurg Psychiatry. 2010;81(11):1243. doi:10.1136/jnnp.2009.197962 PubMed | Crossref | Google Scholar
- Rosenbloom MH, Atri A. The evaluation of rapidly progressive dementia. Neurologist. 2011;17(2):67. doi:10.1097/NRL.0b013e31820ba5e3 PubMed | Crossref | Google Scholar
- Schneider SA, Bird T. Huntington’s disease, Huntington’s disease look-alikes, and benign hereditary chorea: What’s new? Mov Disord Clin Pract. 2016;3(4):342-354. doi:10.1002/mdc3.12312 PubMed | Crossref | Google Scholar
- Gul F, Manzoor H, Aman B, Aman M, Hameed A. Creutzfeldt-Jakob disease: A case report and differential diagnoses. Ann Case Rep. 2023;8:1282. Crossref | Google Scholar
- Gozke E, Erdal N, Unal M. Creutzfeldt-Jacob disease: A case report. Cases J. 2008;1:146. doi:10.1186/1757-1626-1-146 PubMed | Crossref | Google Scholar
- R S, Sina F, Moudi S. Creutzfeldt-Jakob disease: A case report. Caspian J Intern Med. 2021;12(S2):359-362. doi:10.22088/cjim.12.0.359 PubMed | Google Scholar
Acknowledgments
Not applicable
Funding
Not applicable
Author Information
Corresponding Author:
Sheetal Hiremath
Independent Researcher, Department of Content
medtigo India Pvt Ltd, Pune, India
Email: [email protected]
Co-Author:
Rupashi Mukhia
Department of Outcomes Research
Anesthesiology Institute, Cleveland Clinic, USA
Authors Contributions
All patient-related data were collected by Dr. Rupashi Mukhia. Dr. Sheetal Hiremath contributed to the writing of the manuscript, including the original draft preparation and subsequent review and editing to refine the final version.
Informed Consent
Not applicable
Conflict of Interest Statement
This case report is based on a clinical case published in the “Cases” section of medtigo.com. The authors declare no conflicts of interest related to this publication.
Guarantor
Not applicable
DOI
Cite this Article
Hiremath S, Mukhia R. Creutzfeldt-Jakob Disease: A Case Report and Review. medtigo J Med. 2023;1(3):e3062131. doi:10.63096/medtigo3062131 Crossref













