






 Author Affiliations
Author Affiliations
Abstract
Creutzfeldt-Jacob disease (CJD) is a rare, fatal neurodegenerative disorder caused by prion protein accumulation. The Heidenhain variant is an even rarer form, primarily presenting with early visual disturbances, often leading to delayed diagnosis. This case report describes an elderly female who initially presented with visual disturbances, which progressed to severe neurological decline. The initial misdiagnosis of a stroke delayed the recognition of the Heidenhain variant of CJD. Diagnosis was ultimately confirmed by cerebrospinal fluid (CSF) analysis using the real-time quaking-induced conversion (RT-QuIC) assay, and palliative care was provided. This case emphasizes the need for early recognition of prion diseases in patients presenting with visual symptoms and rapidly progressive neurological decline.
Keywords
Creutzfeldt-Jacob disease, Heidenhain variant, Cerebrospinal fluid, Neurodegenerative disorder, Real-time quaking-induced conversion (RT-QuIC) assay.
Introduction
CJD is a rare, transmissible, and fatal neurodegenerative condition caused by the accumulation of misfolded prion proteins in the brain. The disease is characterized by rapidly progressive cognitive and motor dysfunction, typically leading to death within months of onset. Sporadic CJD is the most common form, but it can present atypically in certain variants.[1]
The Heidenhain variant of CJD accounts for approximately 10% of sporadic cases and is characterized by early visual disturbances, such as cortical blindness, hallucinations, and visual field defects.[2] This presentation, involving the occipital cortex early in the disease process, frequently leads to misdiagnosis, such as stroke or psychiatric illness, delaying accurate diagnosis.[3]
In this case report, we describe an old female in her late 70’s who presented with early visual symptoms, which progressed rapidly to cognitive decline and motor dysfunction. The eventual diagnosis of the Heidenhain variant of CJD was confirmed through advanced imaging and CSF analysis.
Case Presentation
Patient demographics: An elderly female in her late 70’s with a known history of hypertension and bilateral cataracts presented to the emergency department (ED) with a one-week history of peripheral double vision and confusion, specifically mixing up names.
Initial presentation and examination: On admission, the patient was found to have normal extraocular movements, muscle tone, reflexes, and sensation, and no focal neurological deficits were identified. Despite her visual complaints, she had no motor or sensory impairment. Initial investigations included a computed tomography (CT) scan of the brain, which was normal, and a magnetic resonance imaging (MRI) that revealed a small area of cortical hyperintensity in the left parietal region (Figure 1).
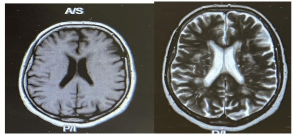
Figure 1: MRI brain showing a small area of parietal cortical hyperintensity
Treatment and initial misdiagnosis: The patient was initially diagnosed with a possible stroke and was started on dual antiplatelet therapy (aspirin and clopidogrel) and a high-dose statin. The ophthalmology and psychiatry teams reviewed the case, and no intervention was suggested for her visual complaints. Given her anxiety and disorientation, anxiolytics were prescribed, considering it a post-stroke stress disorder. The presentation was not compatible with the initial diagnosis, and the level of extreme anxiety was confounding the initial picture. Further evaluation in the stroke radiological meeting by reviewing images and clinical picture ruled out the presence of acute brain ischemia, as was initially considered.
Progression of symptoms: The patient responded well to physiotherapy and occupational therapy initially in the acute stroke unit. Due to her visible disorientation, she was transferred to a post-acute rehabilitation unit for further evaluation and management. During rehabilitation, the patient’s neurological status rapidly declined. She exhibited bizarre behavior, increased disorientation, and abnormal eye movements. Communication became increasingly difficult, and she developed left-sided neglect, and she started having periods of jerky movements. Her condition worsened over three weeks, leading to intermittent irresponsibility and inexpressiveness. In the meantime, patients experienced coffee-ground vomitus, suggesting upper gastrointestinal bleeding. A drop in hemoglobin was noted, and endoscopy revealed a large gastric ulcer. Anticoagulation was stopped.
Further investigations: A repeat MRI revealed bilateral discrete periventricular and subcortical white matter hyperintensities, an electroencephalogram (EEG) showed a typical repetitive pattern of bilateral synchronous periodic epileptiform discharges, and CSF analysis via lumbar puncture revealed positive results for RT-QuIC, confirming the presence of prion protein aggregates. This led to a diagnosis of the Heidenhain variant of CJD.
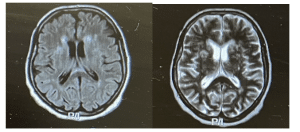
Figure 2: MRI brain showing periventricular hyperintensities

Figure 3: EEG showing periodic sharp wave complexes
Diagnosis: The diagnosis was confirmed as the Heidenhain variant of sporadic CJD, based on clinical progression, EEG findings, MRI findings, and positive RT-QuIC results.[4]
Case Management
The patient was initially started on ceftriaxone, Keppra, acyclovir, intravenous immunoglobulin (IVIG), and steroids, considering worsening encephalopathy vs status epilepticus vs CJD, but these were all discontinued ultimately once CJD was confirmed.
The patient was referred to the palliative care team for symptomatic management and was transferred to hospice care, as no curative treatment exists for CJD. Despite supportive measures, the patient’s condition continued to deteriorate. The family was kept fully informed of her prognosis. Given the confirmed diagnosis via MRI, EEG, and CSF analysis, a post-mortem examination was deemed unnecessary.
Discussion
The Heidenhain variant of CJD presents significant diagnostic challenges due to its atypical initial manifestation with visual disturbances. These visual symptoms are a result of early occipital cortex involvement, which is uncommon in other CJD variants.[5] As in this case, the initial misdiagnosis of stroke delayed the recognition of the underlying prion disease, which is often the case in Heidenhain variant presentations.
The rapid progression of the patient’s condition, characterized by increasing neurological decline, including left-sided neglect and bizarre behavior, was more consistent with prion disease than cerebrovascular events. MRI findings, specifically the diffusion-weighted imaging abnormalities, along with positive RT-QuIC results from the CSF, were essential in making the diagnosis. RT-QuIC, a sensitive assay for detecting prion proteins, has revolutionized the diagnostic approach to CJD.[6]
This case also highlights the importance of a multidisciplinary approach, involving neurology, ophthalmology, psychiatry, and palliative care teams. Early diagnosis can allow for the cessation of unnecessary medications, such as antibiotics and anticoagulants, as seen in this case, thus avoiding potential iatrogenic complications.[7]
Conclusion
The Heidenhain variant of CJD is a rare form of prion disease that requires a high index of suspicion, particularly when patients present with early visual disturbances and rapidly progressive neurological decline. This case underscores the importance of considering prion diseases as a differential diagnosis when faced with unexplained visual symptoms, particularly in elderly patients. Advanced diagnostic tools, such as MRI and RT-QuIC assays, have significantly improved the accuracy of in-vivo diagnosis, reducing reliance on post-mortem confirmation.
Given the absence of curative treatment, early recognition and palliative care involvement are crucial for optimizing patient comfort and supporting families through this devastating illness.
The Heidenhain variant serves as a reminder that prion diseases, though rare, should remain part of the differential diagnosis in cases of rapidly progressing neurological decline, especially when visual disturbances are prominent. Future advancements in prion research may offer hope for better therapeutic interventions, but until then, early diagnosis remains crucial for appropriate management and care.
References
- Zerr I, Kallenberg K, Summers DM, et al. Updated clinical diagnostic criteria for sporadic Creutzfeldt-Jakob disease. 2009;132(10):2659-2668. doi:10.1093/brain/awp191 PubMed | Crossref | Google Scholar
- Parker SE, Gujrati M, Pula JH, Zallek SN, Kattah JC. The heidenhain variant of Creutzfeldt-Jakob disease–a case series. J Neuroophthalmol. 2014 Mar;34(1):4-9. doi: 10.1097/WNO.0b013e3182916155 PubMed | Crossref | Google Scholar
- Manix M, Kalakoti P, Henry M, et al. Creutzfeldt-Jakob disease: Updated diagnostic criteria, treatment algorithm, and the utility of brain biopsy. Neurosurg Focus. 2015;39(5):E2. doi:10.3171/2015.8.FOCUS15328 PubMed | Crossref |
Google Scholar - Hamlin C, Puoti G, Berri S, et al. A comparison of tau and 14-3-3 protein in the diagnosis of Creutzfeldt-Jakob disease. Neurology. 2012;79(6):547-552. doi:10.1212/WNL.0b013e318263565f PubMed| Crossref | Google Scholar
- Geschwind MD. Rapidly progressive dementia: Prion diseases and other rapid dementias. Continuum (Minneap Minn). 2016;22(2):510-537. doi:10.1212/CON.0000000000000306 PubMed
- Appleby BS, Appleby KK, Crain BJ, et al. Characteristics of established and proposed sporadic Creutzfeldt-Jakob disease variants. Arch Neurol. 2009;66(2):208-215. doi:10.1001/archneurol.2008.565 PubMed
- Hermann P, Appleby B, Brandel JP, Caughey B, Collins S, Geschwind MD, Green A, Haïk S, Kovacs GG, Ladogana A, Llorens F, Mead S, Nishida N, Pal S, Parchi P, Pocchiari M, Satoh K, Zanusso G, Zerr I. Biomarkers and diagnostic guidelines for sporadic Creutzfeldt-Jakob disease. Lancet Neurol. 2021 Mar;20(3):235-246. doi: 10.1016/S1474-4422(20)30477-4 PubMed | Crossref | Google Scholar
Acknowledgments
I would like to express my heartfelt gratitude to Dr. Jose Miranda, my consultant, for his invaluable guidance and unwavering support throughout the preparation of this case report. His extensive knowledge and expertise in the field were instrumental in shaping my understanding of the case and the relevant literature. Dr. Miranda’s thoughtful insights and constructive feedback during the writing process greatly enhanced the clarity and quality of my work. His encouragement and mentorship motivated me to approach this report with dedication and rigor. I am sincerely thankful for the time he invested in reviewing my drafts and discussing various aspects of the case, which helped me refine my analysis and conclusions. This report would not have been possible without his support, and I deeply appreciate his commitment to fostering my growth as a clinician.
Funding
This report received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Author Information
Corresponding Author:
Muhammad Kazim Bashir
Department of General Medicine
Letterkenny University Hospital, Ireland
Email: kazimsahab97@gmail.com
Co-Author:
Jose Miranda
Department of Internal Medicine
Letterkenny University Hospital, Ireland
Authors Contributions
All authors contributed to the conceptualization, investigation, and data curation by acquiring and critically reviewing the selected articles. They were collectively involved in the writing – original draft preparation, and writing – review & editing to refine the manuscript. Additionally, all authors participated in the supervision of the work, ensuring accuracy and completeness. The final manuscript was approved by all named authors for submission to the journal.
Ethical Approval
This case report was conducted in accordance with institutional guidelines. As the patient was unable to provide consent, permission to proceed with the publication was obtained from the patient’s sister. All identifying information has been anonymized to protect the patient’s privacy. Ethical approval was not required for this case report, as it adheres to the guidelines for reporting anonymized clinical information.
Informed Consent
Informed consent was obtained from the patient’s sister, as the patient was unable to provide consent due to cognitive impairment. The sister was provided with detailed information regarding the case report, including its purpose and the implications of participation, and she consented on behalf of the patient.
Conflict of Interest Statement
Authors declare no conflict of interests.
Guarantor
None
DOI
Cite this Article
Bashir MK, Miranda J. Atypical Presentation of Heidenhain Variant of Sporadic Creutzfeldt-Jakob Disease: Diagnostic Challenges and Clinical Insight. medtigo J Neurol Psychiatr. 2024;1(1):e3084116. doi:10.63096/medtigo3084116 Crossref













