






 Author Affiliations
Author Affiliations
Abstract
Evans syndrome (ES) is a rare and chronic autoimmune disorder characterized by the simultaneous presence of autoimmune hemolytic anemia (AIHA) and immune thrombocytopenic purpura (ITP), identified by a positive direct antiglobulin test. It is classified into primary and secondary types, with an incidence of 37%–73% in autoimmune hemolytic anemia patients. Clinical presentations encompass jaundice, pallor, mucosal bleeding, and fatigue with remissions and exacerbations during an individual’s lifetime. Treatment lacks established evidence-based protocols, with steroids serving as the first-line therapy. IVIG is employed as a life-saving intervention in cases of chronic immune thrombocytopenic purpura. Second-line treatments for ES include rituximab, cyclosporine, thrombopoietin receptor agonists, mycophenolate mofetil (MMF), sirolimus, azathioprine, and vincristine. Hematopoietic stem cell transplantation has demonstrated success in cases resistant to immunosuppressive drugs; however, its severe side effects need to be carefully considered. In conclusion, ES presents a complex and heterogeneous course, posing challenges to both patients and physicians. Prospective clinical trials are essential to explore potential targeted therapies for achieving improved long-term responses or even a cure.
Keywords
Evans syndrome, Thrombocytopenic purpura, Anemia, Immunoglobulin, Treatment, Supportive care.
Introduction
ES is an uncommon autoimmune condition where the immune system attacks the body’s red blood cells, white blood cells, and/or platelets, leading to their destruction. It is also known as AIHA and autoimmune thrombocytopenia. Dr. Robert Evans initially documented ES in 1951 [1]. ES-anaemia is an AIHA affected by IgG antibodies, and rarely by IgA, thereby ruling out the association of cold agglutinins [2]. Autoimmune neutropenia (AIN) is a component of ES, showing in 15% of cases in adults and 20% in children [3]. In this context, ES was categorized as primary when no associated diagnoses were identified, and secondary when ES coexisted with a concurrent hematological malignancy, rheumatic disorder, or another specific
disease [3].
This literature review explained different modules such as ES epidemiology, signs, symptoms, causes, diagnosis and differential diagnosis, types, and management.
Epidemiology
According to the Office of Rare Diseases (ORD) at the National Institute of Health (NIH), ES is classified as a rare/uncommon disease by the signifying its occurrence in fewer than 200,000 individuals within the U.S. population [4]. A singular study has attempted to quantify its incidence and prevalence in adults. According to data from Denmark in 2016, the annual incidence was 1.8 per 1,000,000 person-years, and the prevalence stood at 21.3 per 1,000,000 living persons. In pre-pubertal children, the estimated incidence ranges between 0.7 and 1.2 per 1,000,000 person-years [5]. ES exhibited a higher prevalence in boys than in girls, with a sex ratio of 2:1. This differs from the typical distribution of autoimmune diseases in adulthood, where women are more common than men [6]. French studies concluded that the annual incidence of 10 in French children under 18 years of age. This corresponds to an approximate annual incidence of 0.7 per 1,000,000 [7].
Signs and Symptoms
The symptoms and severity of ES vary widely among individuals, including differences in onset, course, and duration. Most people encounter a chronic pattern with periods of exacerbated symptoms and temporary remissions generated by treatment. Some individuals may initially present with accelerated destruction of red blood cells, leading to anemia. Symptoms of anemia include fatigue, pallor, lightheadedness, shortness of breath, dark urine, and a rapid heartbeat. Yellowing of the skin and eyes (jaundice) may also occur [8]. Others may exhibit thrombocytopenia, characterized by tiny reddish or purple skin spots (petechiae), larger purplish discolorations (ecchymosis), and a rash (purpura) resulting from internal bleeding [9]. Neutropenia, or low white blood cell levels, is less common in ES. accompanied by symptoms such as fever, malaise, and mouth ulcers. Additional symptoms can include enlarged lymph nodes, spleen, and liver, which may appear intermittently or during acute episodes. Unfortunately, some individuals with ES may be refractory to treatment, leading to critical complications like sepsis, severe bleeding episodes, and heart failure [10].
Causes
The precise underlying cause of ES remains unknown, though it is classified as an autoimmune disorder. In this condition, the immune system produces antibodies that wrongly target healthy tissues, exclusively red blood cells, platelets, and sometimes certain white blood cells [11]. Researchers propose that an initiating event, such as an infection or an essential disorder, may trigger the immune system to make autoantibodies in this disorder. Secondary ESis are associated with various conditions such as Immunoglobulin A (IgA) deficiency, Sjogren’s syndrome, autoimmune lymphoproliferative syndrome (ALPS), lupus, antiphospholipid syndrome, chronic lymphocytic leukemia, certain lymphomas, and common variable immunodeficiency [12].
Pathogenesis
ES might signify a malfunction in the control of immune regulation, encompassing abnormalities in both cellular and humoral immunity. Some individuals with ES have been reported to exhibit decreased helper T-suppressor and increased T-suppressor lymphocytes, resulting in a reduced ratio of CD4/CD8 cells. Additionally, abnormalities in immunoglobulin levels have been observed in certain ES patients [13]. In certain cases, the pathogenesis of ES involves a diminished control of autoreactive B lymphocyte clones by T lymphocytes. This is characterized by an aberrant T helper cell type 1/ T helper cell type 2 (Th1/Th2) ratio, showing increased production of Interleukins – 10 (IL-10) and Interferons (IFN-γ), along with complete suppression of Transforming growth factor-β (TGF-β) [14]. The genomic era has brought forth new insights into the biology of diseases. Stepensky P et al documented that two siblings with ES, viral infections, and progressive leukopenia with a homozygous frame-shift mutation in tripeptidyl peptidase II, resulting in the elimination of its expression [15].
Diagnosis
The diagnosis of ES depends on the concomitant or sequential diagnosis of autoimmune cytopenia (AIC), and the delay between events of AIC is not a restricting factor. The recommended diagnosis details are mentioned in Table 1 [16,17,18]. Diagnostic methodology for ES.
| Type of ES | Diagnosis procedure |
| Primary ES |
|
| Secondary ES
(exclude differential diagnosis) |
|
Table 1: Recommended diagnosis tests for ES
Differential Diagnosis
Disorders that Show Similar Symptoms of ES
Various conditions may exhibit both hemolytic anemia and thrombocytopenia, such as paroxysmal nocturnal hemoglobinuria (PNH), acquired thrombotic thrombocytopenic purpura, hemolytic-uremic syndrome, and Kasabach-Merritt syndrome. ALPS stands as a rare genetic disorder with overlapping features with ES. ALPS is characterized by elevated numbers of lymphocytes, a type of white blood cell, which may accumulate in lymph nodes, liver, and spleen, leading to organ enlargement. The symptoms of ALPS often mirror those of ES, including anemia, thrombocytopenia, and neutropenia. Most individuals with ALPS carry a mutation in the tumor necrosis factor receptor gene superfamily member (TNFRSF6), also known as CD95 or Fas. The precise relationship between these disorders remains incompletely understood. It is believed that about half of children diagnosed with ES may have ALPS as an underlying cause, attributed to the failure of auto-reactive cells to undergo normal programmed cell death (apoptosis). However, the connection between ALPS and ES in adults is less distinctly defined [5,10].
ES Types
The classification of ES includes primary/idiopathic, which is diagnosed by exclusion, indicating no underlying requirement, and secondary when there is a baseline syndrome present. Studies have shown that when compared to primary ES, secondary ES typically responds better.
1. ES during Childhood: Between 1981 and 1990, the incidence of ES in children rose from 0.5 cases per million person-years to 1.2 cases per million person-years between 2006 and 2015. Similarly, the prevalence exhibited a significant increase, escalating from 6.7 cases per million persons in 1990 to 19.3 cases per million in 2015. These figures highlight a notable similarity in both the incidence and prevalence of ES between children and adults. In the pediatric population, ES accounts for 11.7% of isolated AIHA cases and 0.7% of isolated ITP cases [5].
The ES is more common during childhood. For example, Pui CH et al reported that 4.1% of 179 children had ES compared to 0.78% of 766 adults. It’s crucial to highlight that autoimmune cytopenias in children are often linked to primary immunodeficiencies, and ruling out these conditions is essential in this age group [19]. Multicenter retrospective study was carried out across 21 hospitals, and 42 children (median age of 7.7 years) were included. Treatment modalities varied and encompassed 1–12 interventions, with the predominant approach being a combination of steroids and IVIG. Some patients underwent more interventions, such as splenectomy, and/or received medications like cyclosporine, vincristine, danazol, azathioprine, cyclophosphamide, and plasmapheresis. Unfortunately, no optimal response was observed. Complications were observed in 24 (58%) patients, with 12 patients had hemorrhagic complications and the other 12 patients were suffering from severe infections, primarily sepsis, pneumonia, meningitis, abscess, and osteomyelitis [20].
Among the 156 children in the French cohort, 69% children required at least one second-line treatment, with nearly half needing multiple second-line therapies. Initial treatments included steroids and IVIG for ES thrombocytopenia, and steroids for ES anemia. The primary second-line therapies comprised 31% rituximab, 15% azathioprine, 12% splenectomy, 9% cyclosporin, and 2.5% MMF. At final follow-up, children achieved a complete response in about 74%, while 10% of children died because of infections (70%), often exacerbated by ES treatments. For those whose death was ES-related, 30% uncontrolled thrombocytopenia leading to bleeding was the predominant cause [7].
2. ES during Pregnancy: ES during pregnancy is rare, and typically, the diagnosis is established beforehand. In this context, the main differential diagnoses include thrombotic thrombocytopenic purpura, hemolysis, elevated liver enzymes, and low platelets (HELLP) syndrome, and uremic hemolytic syndrome. Notably, maternal IgG antibodies passively transfer to the fetal circulation via the placenta. This phenomenon explains the transient thrombocytopenia or hemolytic anemia that has been reported in fetuses or newborns of autoimmune cytopenia women [21]. When timely and appropriate treatment is given, pregnant women with ES typically have a good outcome. Nonetheless, there’s a higher chance of complications like postpartum hemorrhage and abruptio placentae occurring. The prognosis for fetuses is usually rarer positive, with high-risk complications including severe hemolytic anemia or hemorrhages due to substantial thrombocytopenia, often leading to intracranial bleeding, intra- or extrauterine death, or neurological impairments. Managing hematological therapy during pregnancy is challenging due to the potential teratogenic effects of certain drugs. The preferred treatment is a combination of steroids and IVIG. These treatments function by decreasing the number of antibodies that cross the placenta and by downregulating the mother’s IgG antibodies, which lowers the amount of these antibodies in the bloodstream of the fetus. In refractory cases during the third trimester, the last treatment options may include azathioprine or splenectomy [22,23].
Clinical Trials
Certain individuals with ES have undergone treatment involving allogeneic or autologous stem cell transplantation. Stem cells, specialized cells present in bone marrow, play a crucial role in generating various blood cell types, including red blood cells, white blood cells, and platelets. In autologous stem cell transplantation, the individual’s own stem cells are extracted after prior treatment, typically involving drugs [10]. These healthy stem cells are then reintroduced into the bone marrow once the disorder has been addressed. On the other hand, allogeneic stem cell transplantation involves stem cells donated by another person, often a closely matched family member. These transplantation procedures are generally considered a last resort for individuals who have not responded to other treatment modalities. The outcomes have been variable, and further experience is required to determine their potential effectiveness as a treatment for ES [5].
Management
Managing ES remains a challenge. The syndrome is characterized by periods of remission and exacerbation, and the response to treatment varies, even within the same individual. Management includes first-line, second-line, and third-line therapy.
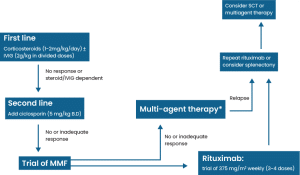
Figure 1: Management of ES
1. First-line therapy: Corticosteroids and IVIG are widely used drugs for first-line management of ES. Firstly, initiate treatment with steroids, mainly prednisolone/methylprednisolone, and consider incorporating IVIG if patients do not show improvement or if they become dependent on steroids [10]. For individuals exhibiting a partial response, the initial prednisolone dose should be sustained for an additional two consecutive weeks. If no improvement is observed, initiation of second-line therapy becomes obligatory. A decline in response is primarily linked to steroid dose reduction or viral infections. IVIG may be introduced if ITP is predominant. Notably, in situations where thrombocytopenia is prominent, a favorable response has been associated with the presence of hepatomegaly [24].
2. Second-line therapy: Second-line treatment options encompass immunosuppressive agents such as cyclosporin, MMF, and danazol, along with the monoclonal antibody rituximab and chemotherapy involving vincristine. Additionally, splenectomy may be regarded as a potential second-line intervention [10]. Michel et al conducted a retrospective multicenter cohort study with 68 ES adults and reported that 11 patients were taken rituximab at the standard dose, after 12 months of follow-up showed 82% response, and 2 of 11 patients were relapsed [3]. The use of MMF has been associated with a heightened risk of developing lymphoma. A prospective study involving 200 patients revealed that the primary adverse effects were epigastric pain and headache. Notably, there were no indications suggestive of lymphoma. The recommended regimen involves administering MMF 600 mg/m2 dose 2 times per day, with a follow-up evaluation three months after the initial treatment due to the risk of relapse and limited information on long-term adverse effects. These findings suggest that MMF can serve as a useful and safe second-line treatment for patients with ES who are refractory to steroids or IVIG [25].
Cyclosporine has been used in ES patients since 1994. If cyclosporine is administered, continuous monitoring of serum concentrations is imperative because of the chronic complications linked to its use. These adverse effects, including malignancy and nephrotoxicity, have been observed even with low doses of cyclosporine (6 mg/kg/day) after 20 years of treatment [26]. Before the advent of rituximab, splenectomy was the sole recourse for refractory relapsing patients. It was traditionally regarded as a second-line treatment for individuals with autoimmune cytopenias, attaining response rates of 60%–75% in AIHA and ITP. However, in the context of ES, the documented responses are notably diverse, ranging between 0% and 66%. [27]. In recent times, rituximab has replaced splenectomy as a preferred treatment option, primarily due to the inherent risks associated with the surgical procedure. It is noteworthy to emphasize that in children under the age of 6, splenectomy is not recommended due to an elevated risk of septicemia/meningitis [28]. Laparoscopic splenectomy presents itself as an alternative that may mitigate certain secondary adverse effects, as evidenced by five patients with ES who underwent this procedure. Among these cases, two achieved normalization of platelet counts at an average of 18 months without requiring further treatment. One patient initially responded well but needed additional medical intervention after 18 months, while two cases did not exhibit a response to laparoscopic splenectomy [27].
3. Third-line therapy: Many patients tend to respond to first or second-line therapy, often maintaining positive outcomes for many years. Nevertheless, for individuals grappling with severe and recurrent disease despite second-line interventions, the exploration of additional options becomes necessary. Primary among the third-line choices are cyclophosphamide, alemtuzumab, or stem cell transplantation (SCT). In the case of older adults, SCT may not be an appealing option due to its relatively high mortality and failure rate in ES, as elaborated below. However, for children experiencing chronically relapsing and life-threatening ES, SCT from a human leukocyte antigen (HLA)-identical donor is likely the most viable avenue for achieving long-term remission, making it superior to other third-line treatments [29]. An adult with refractory ES (ES), resistant to multiple interventions, underwent an allogeneic hematopoietic stem cell transplant (HSCT) with a conditioning regimen consisting of a 200mg per kg dose of cyclophosphamide and a 90mg per kg dose of anti-thymocyte globulin. Three months post-transplant, antibodies against erythrocytes and platelets were negative, indicating a successful response, and the patient achieved 100% chimerization. Remarkably, this individual maintained complete remission throughout the 30 months of follow-up, highlighting the efficacy of allogeneic HSCT in managing refractory cases of ES [30].
Supportive Care
Supportive care is essential for stabilizing patients with ES before proceeding with further treatment. Different care methods include blood and platelet transfusions, Intravenous Fluids, nutritional Support, Counseling, and Psychological Support [10,31].
Complications
The median survival of patients with ES has shown an increase over time, reaching approximately 7 years. Notably, the survival is less favorable in secondary ES than primary ES, with median survival times of 1.7 years for secondary ES and 10.9 years for primary ES. Significantly, 30% of deaths among individuals with ES occur within the first year of diagnosis. The adjusted hazard ratio of death is reported as 12.7 at 1 year, 2.3 between 1–5 years, and 1.5 between 5–10 years, underscoring a higher mortality risk in the initial period following diagnosis [32]. While life-threatening complications are uncommon in ES, their prompt recognition and management are crucial. Cases of ES-thrombocytopenia leading to life-threatening hemorrhage, such as intracranial, visceral, or gastrointestinal bleeding causing profound anemia, should be addressed following the recommendations outlined for isolated ITP. It is important to note that these recommendations are derived from expert committee reports, opinions, or clinical experiences and are categorized as grade C. Despite its delayed onset of action, taking 3 to 4 weeks, early administration of rituximab should be considered in life-threatening conditions to improve further and alleviate serious conditions. In rare instances, emergency splenectomy may be considered, and a typically swift response is often observed in the days following the surgical procedure [33].
Conclusion
ES is a rare disorder characterized by a diverse and often relapsing course throughout an individual’s lifetime, despite employing various treatment approaches. The interplay of genetic and epigenetic factors is believed to contribute to its pathogenesis. Understanding the precise etiology of the disease holds the key to developing targeted therapies with fewer adverse effects, thereby significantly enhancing the quality of life for affected individuals. Long-term survival data for ES are limited, with patients being tracked over a median range of 3 to 8 years. During this period, mortality rates varied between 7% and 36%. The primary causes of death were attributed to hemorrhage and sepsis. Finally, it was concluded that it is essential for researchers to understand ES’s pathophysiology and improve long-term treatment effectiveness by conducting various clinical trials.
References
- vans RS, Takahashi K, Duane RT, Payne R, Liu C. Primary thrombocytopenic purpura and acquired hemolytic anemia: evidence for a common etiology. AMA Arch Intern Med. 1951;87(1):48-65. doi:10.1001/archinte.1951.03810010058005 PubMed | Crossref | Google Scholar
- Moncharmont P, Troncy J, Rigal D. IgA anti-red blood cell auto-antibodies in ES. Hematology. 2007;12(6):587-589. doi:10.1080/10245330701521481 PubMed | Crossref | Google Scholar
- Michel M, Chanet V, Dechartres A, et al. The spectrum of ES in adults: new insight into the disease based on the analysis of 68 cases. Blood. 2009;114(15):3167-3172. doi:10.1182/blood-2009-04-215368 PubMed | Crossref | Google Scholar
- Karakantza M, Mouzaki A, Theodoropoulou M, Bussel JB, Maniatis A. Th1 and Th2 cytokines in a patient with Evans’ syndrome and profound lymphopenia. Br J Haematol. 2000;110(4):968-970. doi:10.1046/j.1365-2141.2000.02296.x PubMed | Crossref | Google Scholar
- Mannering N, Hansen DL, Frederiksen H. ES in children below 13 years of age – A nationwide population-based cohort study. PLoS One. 2020;15(4):e0231284. doi:10.1371/journal.pone.0231284 PubMed | Crossref | Google Scholar
- Rose NR. Prediction and prevention of autoimmune disease in the 21st century: review and preview. Am J Epidemiol. 2016;183(5):403-406. doi:10.1093/aje/kwv292 PubMed | Crossref | Google Scholar
- Aladjidi N, Fernandes H, Leblanc T, et al. ES in children: long-term outcome in a prospective French national observational cohort. Front Pediatr. 2015;3:79. doi:10.3389/fped.2015.00079 PubMed | Crossref | Google Scholar
- ES. Symptoms & Causes. Boston Children’s Hospital. November 27, 2020. Evans syndrome
- Bilochvostenko MA, Ermens AAM, Fiets RB. ES with auto-immune neutropenia. Int J Lab Hematol. 2022;44(1):61-62. doi:10.1111/ijlh.13650 PubMed | Crossref | Google Scholar
- Norton A, Roberts I. Management of ES. Br J Haematol. 2006;132(2):125-137. doi:10.1111/j.1365-2141.2005.05809.x PubMed | Crossref
- Michel M. Adult Evans’ syndrome. Hematol Oncol Clin North Am. 2022;36(2):381-392. doi:10.1016/j.hoc.2021.12.004 PubMed | Crossref | Google Scholar
- Mantadakis E, Farmaki E. Natural history, pathogenesis, and treatment of ES in children. J Pediatr Hematol Oncol. 2017;39(6):413-419. doi:10.1097/MPH.0000000000000897 PubMed | Crossref | Google Scholar
- Wang W, Herrod H, Pui CH, et al. Immunoregulatory abnormalities in ES. Am J Hematol. 1983;15:381-390. doi:10.1002/ajh.2830150409 PubMed | Crossref | Google Scholar
- Karakantza M, Mouzaki A, Theodoropoulou M, et al. Th1 and Th2 cytokines in a patient with Evans’ syndrome and profound lymphopenia. Br J Haematol. 2000;110:968-970. doi:10.1046/j.1365-2141.2000.02296.x PubMed | Crossref | Google Scholar
- Stepensky P, Rensing-Ehl A, Gather R, et al. Early-onset ES, immunodeficiency, and premature immunosenescence associated with tripeptidyl-peptidase II deficiency. Blood. 2015;125:753-761. doi:10.1182/blood-2014-08-593202 PubMed | Crossref | Google Scholar
- Jager U, Barcellini W, Broome CM, et al. Diagnosis and treatment of autoimmune hemolytic anemia in adults: recommendations from the first international consensus meeting. Blood Rev. 2020;41:100648. doi:10.1016/j.blre.2019.100648 PubMed | Crossref | Google Scholar
- Provan D, Arnold DM, Bussel JB, et al. Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood Adv. 2019;3(22):3780-3817. doi:10.1182/bloodadvances.2019000812 PubMed | Crossref | Google Scholar
- Porcelijn L, Huiskes E, Oldert G, et al. Detection of platelet autoantibodies to identify immune thrombocytopenia: state of the art. Br J Haematol. 2018;182(3):423-426. doi:10.1111/bjh.15404 PubMed | Crossref | Google Scholar
- Pui CH, Wilimas J, Wang W. Evans syndrome in childhood. J Pediatr. 1980;97(5):754-758. doi:10.1016/s0022-3476(80)80258-7 PubMed | Crossref | Google Scholar
- Mathew P, Chen G, Wang W. Evans syndrome: results of a national survey. J Pediatr Hematol Oncol. 1997;19(5):433-437. doi:10.1097/00043426-199709000-00005 PubMed | Crossref | Google Scholar
- Sankaran S, Robinson SE. Immune thrombocytopenia and pregnancy. Obstet Med. 2011;4(4):140-146. doi:10.1258/om.2011.110025 PubMed | Crossref | Google Scholar
- Lefkou E, Nelson-Piercy C, Hunt BJ. Evans’ syndrome in pregnancy: a systematic literature review and two new cases. Eur J Obstet Gynecol Reprod Biol. 2010;149(1):10-17. doi:10.1016/j.ejogrb.2009.11.022 PubMed | Crossref | Google Scholar
- Ni H, Chen P, Spring CM, et al. A novel murine model of fetal and neonatal alloimmune thrombocytopenia: response to intravenous IgG therapy. Blood. 2006;107(7):2976-2983. doi:10.1182/blood-2005-06-2562 PubMed | Crossref | Google Scholar
- Ricci S, Lippi F, Canessa C, et al. Efficacy and safety of human intravenous immunoglobulin 5% (Ig VENA) in pediatric patients affected by primary immunodeficiency. Int J Immunopathol Pharmacol. 2020;34:2058738420943006. doi:10.1177/2058738420943006 PubMed | Crossref | Google Scholar
- Miano M, Scalzone M, Perri K, et al. Mycophenolate mofetil and sirolimus as second or further line treatment in children with chronic refractory primitive or secondary autoimmune cytopenias: a single centre experience. Br J Haematol. 2015;171(2):247-253. doi:10.1111/bjh.13533 PubMed | Crossref | Google Scholar
- Janić D, Krivokapić-Dokmanović L, Jovanović N, et al. Glucocorticoid-resistant Evans’ syndrome successfully controlled with low-dose cyclosporine. Int J Clin Pharmacol Ther. 2011;49(10):622-625. Glucocorticoid-resistant Evans’ syndrome successfully controlled with low-dose cyclosporine
- Duperier T, Felsher J, Brody F. Laparoscopic splenectomy for ES. Surg Laparosc Endosc Percutan Tech. 2003;13(1):45-47. doi:10.1097/00129689-200302000-00011 PubMed | Crossref | Google Scholar
- Leone G, Pizzigallo E. Bacterial infections following splenectomy for malignant and nonmalignant hematologic diseases. Mediterr J Hematol Infect Dis. 2015;7(1):e2015057. doi:10.4084/MJHID.2015.057 PubMed | Crossref | Google Scholar
- Chihara D, Sakamoto T, Arimoto-Miyamoto K, et al. Refractory ES after autologous stem cell transplantation for multiple myeloma: management with a second transplantation. Intern Med. 2010;49(7):683-687. doi:10.2169/internalmedicine.49.2922 PubMed | Crossref | Google Scholar
- Oyama Y, Papadopoulos EB, Miranda M, et al. Allogeneic stem cell transplantation for ES. Bone Marrow Transplant. 2001;28(9):903-905. doi:10.1038/sj.bmt.1703237 PubMed | Crossref | Google Scholar
- Miano M. How I manage ES and AIHA cases in children. Br J Haematol. 2016;172(4):524-534. doi:10.1111/bjh.13866 PubMed | Crossref | Google Scholar
- Hansen DL, Möller S, Andersen K, et al. Evans syndrome in adults – incidence, prevalence, and survival in a nationwide cohort. Am J Hematol. 2019;94(10):1081-1090. doi:10.1002/ajh.25574 PubMed | Crossref | Google Scholar
- Provan D, Arnold DM, Bussel JB, et al. Updated international consensus report on the investigation and management of primary immune thrombocytopenia. Blood Adv. 2019;3(22):3780-3817. doi:10.1182/bloodadvances.2019000812 PubMed | Crossref | Google Scholar
Acknowledgments
Not applicable
Funding
Not applicable
Author Information
Bhandhavi Vasireddy
Independent Researcher, Department of Content
medtigo India Pvt Ltd, Pune, India
Email: [email protected]
Author Contribution
The author contributed to the conceptualization, investigation, and data curation by acquiring and critically reviewing the selected articles and was involved in the writing – original draft preparation and writing – review & editing to refine the manuscript.
Informed Consent
Not applicable
Conflict of Interest Statement
Not applicable
Guarantor
Not applicable
DOI
Cite this Article
Vasireddy B. A Study of the Evans Syndrome Literature. medtigo J Med. 2023;1(2):e3062122. doi:10.63096/medtigo3062122 Crossref













